Publication: Unravelling the genomic landscape of Canadian Borrelia burgdorferi: a comparison across global strains
Open Access (free download) - click here.
Authors: Anthony Piot 1,2, Iain L. Mainprize 3, Justin Wood 3 , Jeff Gauthier 1,2, Cezar M. Khursigara 4, Karine Thivierge 5,6, Melanie K.B. Wills 3, and Roger C. Levesque 1,2.
Affiliations:
1 Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Québec City, Québec, Canada;
2 Faculté de médecine, Université Laval, Québec City, Québec, Canada;
3 G. Magnotta Lyme Disease Research Lab, Department of Molecular and Cellular Biology, University of Guelph, Guelph, Canada;
4 Department of Molecular and Cellular Biology, University of Guelph, Guelph, Canada;
5 Laboratoire de santé publique du Québec, Québec, Canada;
6 Institute of Parasitologie, McGill University, Sainte-Anne-de- Bellevue, Canada.
Short Summary
This research explores the genetic composition of Borrelia burgdorferi, the bacteria responsible for Lyme disease, specifically focusing on strains found in Canada. Lyme disease is a growing public health concern in Canada as the ticks that carry the bacteria spread northward due to climate change.
Why This Study is Important
· Filling Knowledge Gaps: Little is currently known about the genetic characteristics and evolutionary history of Canadian B. burgdorferi strains. This lack of information makes it harder to understand how the pathogen originated and adapted to its new environment.
· Providing Genomic Resources: By generating the first complete genome sequences of six Canadian B. burgdorferi strains, our study offers valuable genetic data. These resources will help us and others track the origins and evolution of Lyme disease in Canada, which can inform public health strategies and disease management.
Key Findings of the Research
· Advanced Sequencing Techniques: Reconstructing these genomes is difficult because Borrelia genomes have variable content and structure, even within the same species or strain. This rapid evolution makes it challenging to use existing reference genomes for assembly. This study presents a hybrid approach to ensure the sequence and structure of the genomes are generated accurately.
· Genome Sequencing Output: The study successfully sequenced and assembled the complete genomes (including both the main chromosome and smaller DNA structures called plasmids) of six Canadian B. burgdorferi strains. We then compared these to other Borrelia genomes from the USA and Europe.
· Genome Structure Comparison: The overall structures of the Canadian Borrelia genomes were similar to other B. burgdorferi strains globally. However, we found differences in the content and structure of their plasmids.
· Plasmid Variability: Borrelia species have complex genomes with a main linear chromosome and many smaller linear and circular plasmids. These plasmids are highly variable in number and identity, even among strains of the same species. This variability in plasmid content means that the genes they carry can also differ greatly between strains.
Despite their small size, Borrelia genomes are complex to assemble due to their unique structure and variable plasmid content. This study successfully reconstructed the complete genomes of Canadian B. burgdorferi strains, providing critical insights into their fundamental blueprints. This information is vital for understanding the origins and evolution of Lyme disease in Canada, which will ultimately aid in developing better public health strategies and disease management in newly affected regions.
Deeper Perspectives
Who. This work was a collaborative effort involving teams at Universite Laval in Quebec, and the G. Magnotta Lab at the University of Guelph in Ontario. Dr. Roger Levesque at U. Laval is an internationally regarded scholar in infectious disease genomics. He has led large scale characterization initiatives of other pathogens including salmonella and pseudomonas.
Why: Understanding genomes and biodiversity
Lyme disease is caused by bacteria belonging to the Lyme Borrelia complex, but Borrelia are not identical. Different species are prevalent in different regions of the world (B. burgdorferi is dominant in North America, while B. garinii and B. afzelii are common in Europe). Significant variability below the species level, in strains or isolates, can also influence the disease-causing potential of the bacterium. This pathogenic capacity is determined fundamentally by the genes / genome.
In the simplest terms, individual genes are like recipes in a cookbook (the genome) that holds all the instructions for life. They are written in a chemical language of four DNA bases (A, T, C, G) that we can read and understand using different lab instruments and computational tools. How – and why - scientists read the genome depends on the questions we want to ask. Targeted approaches read individual genes (“recipes”), because it is valuable to understand what the gene encodes, or because we can compare the same gene across different organisms to evaluate their similarities and differences. The latter is a way of understanding biodiversity, and common strategies used for high-level characterization of Borrelia analyze a single gene (OspC typing) or 8 different genes (so called Multi-locus sequence typing; MLST) to classify strains.
Whole genome sequencing (WGS) takes a much more comprehensive approach, and reads all the genes and pieces of DNA between the genes, including genes of unknown function and regions of the DNA that are involved in regulation. Completely solving a genome involves three main steps: sequencing (reading the chemical code), assembling the sequenced fragments into the original order, and annotating the genome to identify the genes, regulatory regions, and other elements that can be characterized.
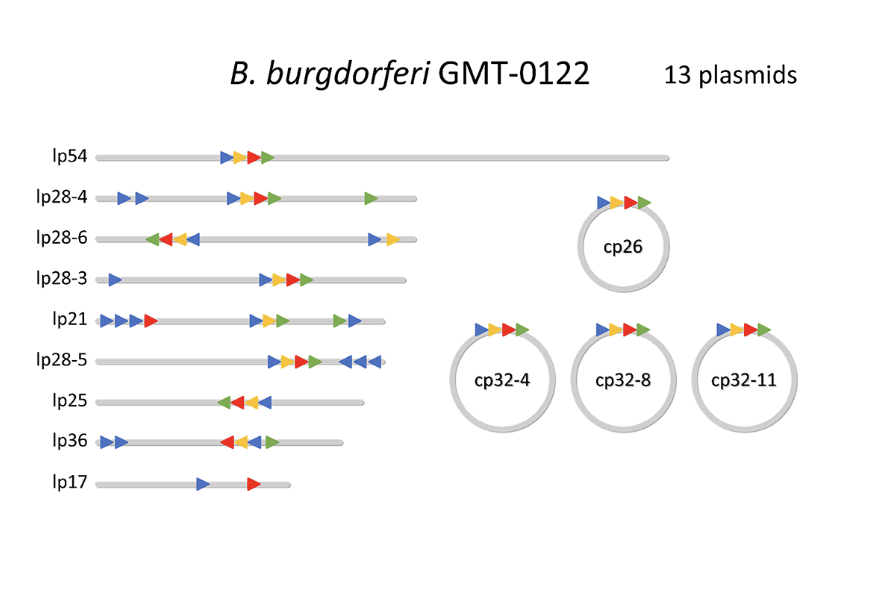
Plasmid content of a strain of Borrelia burgdorferi isolated by the G. Magnotta Lab from a host-attached tick in Ontario, Canada.
The WGS process is more difficult for Lyme Borrelia than for other similar organisms. While most bacteria have a single circular chromosome that contains the majority of the essential genes, Borrelia has the most fragmented bacterial genome currently known to science. The essential components are distributed across a linear chromosome and extrachromosomal genetic elements called plasmids. Borrelia also have a diverse and complex accessory genome, not common to all strains, which is spread across more plasmids. The genomes of these pathogens are considered to be rapidly evolving, as the content (genes, regulatory elements) and the structure (overall blueprint) can look different between closely related species or strains.
Although some studies have reported genome sequences for Canadian strains, they focused on the chromosome and key plasmids. In our study, we reconstructed everything – including the chromosome and all plasmids - for our strains of interest. These included six strains that were isolated from ticks in Canada. This approach provides unparalleled resolution of the ‘recipe book’ of this pathogen and how its genetic capacity can be sculpted by regional variation.
How
Our team developed a processing pipeline that used two independent sequencing platforms to address the complexity of the Lyme disease bacterium. As the name suggests, long-read sequencing can decode longer contiguous sections of DNA, making it easier to put the segments together in the right order (assembly). Historically, this method has been more error-prone at resolving the fine details, so a short-read high-fidelity technique was used to improve the accuracy. Used together, they ensure that the structure and the sequence of the genome are correct. This allowed so called “de-novo” sequencing, which does not use an existing reference genome as a scaffold to construct the new genome. Lack of reliance on known genomes is critical when trying to piece together rapidly evolving genomes like that of Borrelia.
What we found
The two-prong sequencing strategy worked very well to generate complete Borrelia genomes de-novo with high confidence. This was a key outcome as a proof of concept for the approach.
Considering evolutionary relationships, the strains did not cluster by sampling location, meaning that the Canadian strains had various degrees of similarity to strains of American and European origin. The lack of common origin for strains from the same region suggest a complex migration pattern rather than simple Northward expansion from the United States.
Resequencing of the classic lab strain of Borrelia, B31, which is extensively used in research studies, showed widespread plasmid loss when compared against the original sequence first solved in 1997. This reinforces the fluidity of the Borrelia genome and the necessity to work with multiple strains to ensure that lab investigations reflect the true nature and variability of this pathogen.
What’s next
This project has yielded an effective strategy for the comprehensive characterization of Borrelia. Ongoing efforts in the G. Magnotta Lab to isolate more Borrelia strains from large numbers of Canadian ticks will fuel this pipeline and enable deeper insights into the origins and diversity of these pathogens. A key focus of the G. Magnotta Lab is the clinical implications of pathogen biology. New, sequenced isolates will therefore be used in experiments to evaluate novel diagnostic test designs.